
FUSIDIC ACID, 6990-06-3
2-[(1S,2S,5R,6S,7S,10S,11S,13S,14Z,15R,17R)-13-(acetyloxy)-5,17-dihydroxy-2,6,10,11-tetramethyltetracyclo[8.7.0.02,7.011,15]heptadecan-14-ylidene]-6-methylhept-5-enoic acid
Taksta (CEM-102)
Clinical-stage pharmaceutical firm Cempra has secured orphan drug status from the US Food and Drug Administration (FDA) for its drug candidate Taksta (CEM-102) to treat patients with prosthetic joint infections (PJI).
Cempra’s Taksta secures FDA orphan drug status for prosthetic joint infections treatment
TAKSTATM (CEM-102)
Fusidic acid is a bacteriostatic antibiotic that is often used topically in creams and eyedrops, but may also be given systemically as tablets or injections. The global problem of advancing antimicrobial resistance has led to a renewed interest in its use recently.
Fusidic acid acts as a bacterial protein synthesis inhibitor by preventing the turnover ofelongation factor G (EF-G) from the ribosome. Fusidic acid is effective primarily ongram-positive bacteria such as Staphylococcus species, Streptococcus species, and Corynebacterium species. Fusidic acid inhibits bacterial replication and does not kill the bacteria, and is therefore termed bacteriostatic.
Fusidic acid is a true antibiotic, derived from the fungus Fusidium coccineum and was developed by Leo Laboratories in Ballerup, Denmark and released for clinical use in the 1960s. It has also been isolated from Mucor ramannianus and Isaria kogana. The drug is licensed for use as its sodium salt sodium fusidate, and it is approved for use under prescription in South Korea, Japan, UK, Canada, Europe, Australia, New Zealand, Thailand, India and Taiwan. A different oral dosing regimen, based on the compound’s Pharmacokinetic/pharmacodynamic (PK-PD) profile is in clinical development in the U.S. as Taksta.
Fusidic acid (TAKSTATM, CEM-102) is an antibiotic with a long history of safety and efficacy outside the United States. Cempra has exclusive rights to the supply of the compound for the U.S. market. Fusidic acid is orally active against gram-positive bacteria, including all S. aureus strains such as HA-MRSA and CA-MRSA. A novel dosing regimen has been successfully evaluated in a Phase II trial in patients with acute bacterial skin and skin structure infections (aBSSSI). Cempra is conducting a Phase II trial of TAKSTA for patients with prosthetic joint infections.
Profile of TAKSTA (CEM-102)
Prosthetic joint infections (PJI) occur in about 1% of hip replacements and 2% of knee replacements, translating to an incidence rate of about 10,000 per year in the U.S. at current hip and knee arthroplasty rates. There are few good options to treat these serious staphylococcal, often MRSA infections, which require long-term antibiotic treatment. Current therapy in the U.S. is with intravenous antibiotics such as vancomycin. An oral drug that can be safely administered for a long period of time could improve care and quality of life for these patients.
TAKSTA has shown potent activity against a large number of S. aureus strains, including CA-MRSA, HA-MRSA and linezolid-resistant strains, isolated in the U.S over a 10 year period. Its broad S. aureus coverage makes it useful for a broad range of clinical applications. Because of its safety and tolerability profile, TAKSTA could be ideal for patients suffering from staphylococcal infections that require long-term therapy such as patients with PJIs.
Cempra has developed a unique oral loading dose regimen to optimize key pathogen coverage and minimize drug resistance development. This regimen is incorporated in our Phase II trial to treat PJIs with TAKSTA in combination with rifampin, which is commonly used with injectible antibiotics such as vancomycin to treat PJIs.
Research on TAKSTA
Publications
The links for the articles go to subscription-based sites and may require a fee to view the article.
In Vitro Activity of CEM-102 (Fusidic Acid) Against Prevalent Clones and Resistant Phenotypes of Staphylococcus aureus
DF Sahm, J Deane, CM Pillar, P Fernandes
Antimicrobial Agents and Chemotherapy. June 2013 57: 4535-4346
http://aac.asm.org/content/57/9/4535
Efforts to Support the Development of Fusidic Acid in the United States
P Fernandes, D Pereira
Clinical Infectious Disease. June 2011 52:S542-6
http://www.ncbi.nlm.nih.gov/pubmed/21546632
Case report: Treatment of Chronic Osteomyelitis
CR Wolfe
Clinical Infectious Disease. June 2011 52:S538-41
http://cid.oxfordjournals.org/content/52/suppl_7/S538.long
The Safety Record of Fusidic Acid in Non-US markets: A Focus on Skin Infections
CN Kraus, BW Burnstead
Clinical Infectious Disease. June 2011 52:S527-37
http://cid.oxfordjournals.org/content/52/suppl_7/S527.long
A Randomized, Double-Blind Phase 2 Study Comparing the Efficacy and Safety of an Oral Fusidic Acid Loading-Dose Regimen to Oral Linezolid in the Treatment of Acute Bacterial Skin and Skin Structure Infections
JC Craft, SR Moriarty, K Clark, D Scott, TP Degenhardt, JG Still, GR Corey, A Das, P Fernandes
Clinical Infectious Disease. June 2011 52:S520-26
http://cid.oxfordjournals.org/content/52/suppl_7/S520.long
Application of Pharmacokinetic-Pharmacodynamic Modeling and the Justification of a Novel Fusidic Acid Dosing Regimen: Raising Lazarus from the Dead
BT Tsuji, OO Okusanya, JB Bulitta, A Forrest, SM Bhavnani, P Fernandes, PG Ambrose
Clinical Infectious Disease. June 2011 52:S513-19
http://cid.oxfordjournals.org/content/52/suppl_7/S513.long
Pharmacokinetics and Safety of Single, Multiple, and Loading Doses of Fusidic Acid in Healthy Subjects
JG Still, K Clark, TP Degenhardt, D. Scott, P. Fernandes, M. J. Gutierrez
Clinical Infectious Disease. June 2011 52:S504-12
http://cid.oxfordjournals.org/content/52/suppl_7/S504.long
Activity of Fusidic Acid Against Extracellular and Intracellular Staphylococcus aureus: Influence of pH and Comparison with Linezolid and Clindamycin
S Lemaire, F Van Bambeke, D Pierard, PC Appelbaum, PM Tulkens
Clinical Infectious Disease. June 2011 52:S493-503
http://cid.oxfordjournals.org/content/52/suppl_7/S493.long
Characterization of Global Patterns and the Genetics of Fusidic Acid Resistance
DJ Farrell, M Castanheira, I Chopra
Clinical Infectious Disease. June 2011 52:S487-92
http://cid.oxfordjournals.org/content/52/suppl_7/S493.long
In Vitro Antimicrobial Findings for Fusidic Acid Tested Against Contemporary (2008-2009) Gram-Positive Organisms Collected in the United States
RN Jones, RE Mendes, HS Sader, M Castanheira
Clinical Infectious Disease. June 2011 52:S477-86
http://cid.oxfordjournals.org/content/52/suppl_7/S477.long
New Rules for Clinical Trials in Patients with Acute Bacterial Skin and Skin Structure Iinfections: Do not Let the Perfect be the Enemy of the Good
GR Corey, ME Stryjewski
Clinical Infectious Disease. June 2011 52:S469-76
http://cid.oxfordjournals.org/content/52/suppl_7/S469.long
Introduction: Fusidic Acid Enters the United States
RC Moellering, GR Corey, ML Grayson
Clinical Infectious Disease. June 2011 52:S467-8
http://cid.oxfordjournals.org/content/52/suppl_7/S467.long
Evaluation of the Pharmacokinetics-Pharmacodynamics of Fusidic Acid Against Staphylococcus aureus and Streptococcus pyogenes Using In Vitro Infection Models: Implications for Dose Selection
OO Okusanya, BT Tsuji, JB Bulitta, A Forrest, CC Bulik, SM Bhavnani, P Fernandes, PG Ambrose
Diagnostic Microbiology & Infectious Disease. June 2011 70:101-11
http://www.ncbi.nlm.nih.gov/pubmed/21513848
In Vitro Activity of Fusidic Acid (CEM-102, Sodium Fusidate) Against Staphylococcus aureus Isolated from Cystic Fibrosis Patients and its Effect on the Activities of Tobramycin and Amikacin against Pseudomonas aeruginosa and Burkholderia cepacia
P McGhee, K Credito, L Beachel, PC Appelbaum, K Kosowaska-Shick
Antimicrobial Agents and Chemotherapy. June 2011 55:2417-19
http://www.ncbi.nlm.nih.gov/pubmed/21513848
Occurrence and Molecular Characterization of Fusidic Acid Resistance Mechanisms Among Staphylococcus spp. From European Countries (2008)
Castanheira, M., AA Watters, RE Mendes, DJ Farrell, RN Jones
Antimicrobial Agents and Chemotherapy. April 2010 65:1353-8
http://jac.oxfordjournals.org/content/65/7/1353.long
Update on Fusidic Acid (CEM-102) Tested Against Neisseria gonorrhoeae and Chlamydia trachomatis
R Jones, D Biedenbach, P Roblin, S Kohlhoff, M Hammerschlag
Antimicrobial Agents and Chemotherapy. October 2010 54: 4518-4519
http://aac.asm.org/cgi/content/citation/54/10/4518
Fusidic Acid Resistance Rates and Prevalence of Resistance Mechanisms Among Staphylococcus spp. Isolated in North America and Australia, 2007-2008
M Castanheira, AA Watters, JM Bell, JD Turnidge, RN Jones
Antimicrobial Agents and Chemotherapy. September 2010 54: 3614-3617
http://www.ncbi.nlm.nih.gov/pubmed/20566766
Spectrum of Activity, Mutation Rates, Synergistic Interactions, and the Effects of pH and Serum Proteins for Fusidic Acid (CEM-102)
D Biedenbach, P Rhomberg, R Mendes, R Jones
Diagnostic Microbiology & Infectious Disease. March 2010 66: 301-307
http://www.dmidjournal.com/article/S0732-8893(09)00424-6/abstract
Performance of Fusidic Acid (CEM-102) Susceptibility Testing Reagents: Broth Microdilution, Disk Diffusion, and Etest Methods as Applied to Staphylococcus aureus
R Jones, M Castanheira, P Rhomberg, L Woosley, M Pfaller
Journal of Clinical Microbiology. March 2010 48: 972-976
http://jcm.asm.org/cgi/content/abstract/48/3/972
Evaluation of the Activity of Fusidic Acid Tested Against Contemporary Gram-Positive Clinical Isolates From the USA and Canada
M Pfaller, M Castaneira, H Sader, R Jones
International Journal of Antimicrobial Agents. March 2010 35: 282-287
http://www.ijaaonline.com/article/S0924-8579(09)00510-X/abstract
Quantitative and qualitative assessment of antibiotic activity against Staphylococcus aureus biofilm.
Siala, W., M. P. Mingeot-Leclercq, P. M. Tulkens, and F. Van Bambeke.
Abstr. 6th Am. Soc. Microbiol. Conf. Biofilms, abstr A-179.
Download Poster 
Activity of Fusidic Acid Against Methicillin-resistant Staphylococcus Aureus (MRSA) Isolated from CF Patients
Prabhavathi Fernandes, Donald Anderson, K. Kosowska-Shick, P. McGhee, L. Beachel and P.C. Appelbaum
Download Abstract | Download Poster
Evaluation of L6 Ribosomal Protein Alterations in Fusidic Acid-Resistant Staphylococcus aureus: Fitness Cost and Time Kill Analysis
M Castanheira, RN Jones, LN Woosley, RE Mendes, GJ Moet, DJ Farrell
Download Abstract
Fusidic Acid Activity and Coverage of Gram-positive Pathogens Associated with Acute Bacterial Skin and Skin Structure Infections (ABSSSI) in the USA (2008-2010)
RN Jones, DJ Farrell, HS Sader, M Castanheira
Download Abstract | Download Poster
Activity of Fusidic Acid Tested Against Contemporary Staphylococcus aureus Collected from United States Hospitals
M. Castanheira, R.E. Mendes, P.R. Rhomberg, R.N. Jones
Download Abstract | Download Poster
Pharmacokinetics-Pharmacodynamics (PK-PD) of CEM- 102 (Sodium Fusidate) Against Streptococcus pyogenes Using In Vitro Pharmacodynamic Models (IVPM)
B. T. Tsuji, A. Forrest, P. A. Kelchlin, T. Brown, P. N. Holden, O. O. Okusanya, S. M. Bhavnani, P. Fernandes, P. G. Ambrose
Download Abstract | Download Poster
Activity of CEM-102 (sodium fusidate) against 40 MRSA from Cystic Fibrosis Patients
Cynthia Todd, Pamela Mcghee, and Peter Appelbaum
Download Abstract | Download Poster
Ability of CEM-102 (Fusidic Acid), Linezolid, Daptomycin to Select Resistant S.aureus Mutants at Steady-state Serum Levels
K. Kosowska-Shick, P. Mcghee, L. Beachel, P. C. Appelbaum;
Download Abstract | Download Poster
CEM-102 (Fusidic Acid) Maintains Potency against Resistant MRSA and Prevalent Hospital Acquired, Community Acquired,and Epidemic MRSA Clones
C.M. Pillar, M.K. Torres, D.F. Sahm and P. Fernandes
Download Abstract | Download Poster
In Vitro Activity Of Fusicic Acid (CEM-102) Against Resistant Strains Of Staphylococcus aureus
J. dubois, P. Fernandes
Download Abstract | Download Poster
Trade names and preparations
- Fucidin (of Leo in Canada and the US)
- Fucidin H (topical cream with corticosteroid - Leo)
- Fucidin (of Leo in UK/ Leo-Ranbaxy-Croslands in India)
- Fucidine (of Leo in France)
- Fucidin (of Leo in Norway)
- Fucidin (of Adcock Ingram, licenced from Leo, in South Africa)
- Fucithalmic (of Leo in the UK, the Netherlands, Denmark and Portugal)
- Fucicort (topical mixture with hydrocortisone)
- Fucibet (topical mixture with betamethasone)
- Ezaderm (topical mixture with betamethasone)(of United Pharmaceutical “UPM” in Jordan)
- Fuci (of pharopharm in Egypt)
- Fucizon (topical mixture with hydrocortisone of pharopharm in Egypt)
- Foban (topical cream in New Zealand)
- Betafusin (cream mixture with betamethasone valerate in Greece)
- Fusimax (of Schwartz in India)
- Fusiderm (topical cream and ointment by indi pharma in India)
- Fusid (in Nepal)
- Fudic (topical cream in India)
- Fucidin (후시딘, of Dong Wha Pharm in South Korea)
- Stanicid (in Serbia)
- Dermy (Topical cream of W.Woodwards in Pakistan)
- Fugen Cream (膚即淨軟膏 in Taiwan)
- Phudicin Cream (in China; 夫西地酸[24])
- Dermofucin cream ,ointment and gel (in Jordan)
- Optifucin viscous eye drops (of API in Jordan)
- Verutex (of Roche in Brazil)
- TAKSTA (of Cempra in U.S.)
- Futasole (of Julphar in Gulf and north Africa)
- Stanicid (2% ointment of Hemofarm in Serbia)
- Fuzidin (tablets of Biosintez in Russia)
- Fuzimet (ointment with methyluracil of Biosintez in Russia)
- Axcel Fusidic Acid(2% cream and ointment of Kotra Pharma, Malaysia)
MORE INFO

Fusidic acid (FA) is a tetracyclic triterpenoid or fusidane (steroidal) antibiotic derived from the fungus Fusidium coccineum that inhibits bacterial protein synthesis. FA is effective against gram-positive bacteria such as Staphylococcusspecies and Corynebacterium species (L. Verbist, J. Antimicro. Chemo. 25, Suppl. B, 1-5 (1990); A. Bryskier, Fusidic Acid, Chapter 23, in Antimicrobial Agents: Antibacterials and Antifungals (Andre Bryskier, Ed., ASM Press, Washington, USA, 2005)). FA also has moderate activity against Group A beta-hemolytic streptococci, or Streptococcus pyogenes (L. Verbist, J. Antimicro. Chemo. 25, Suppl. B, 1-5 (1990); A. Bryskier, Fusidic Acid, Chapter 23, inAntimicrobial Agents: Antibacterials and Antifungals (Andre Bryskier, Ed., ASM Press, Washington, USA, 2005); Skov et al., Diag. Micro. Infect. Dis. 40:111-116 (2001)).
-
Fusidic acid, chemically (3α, 4α, 8α, 9α, 11α, 13α, 14α, 16α, 17Z)-16-(Acetyloxy)-3,11-dihydroxy-29-nordammara-17(20), 24-dien-21-oic acid, is an antibacterial agent. It is a well-known antibiotic with a unique steroid-like tetracyclic ring system structure, and it is the most potent of a small family of steroidal antibiotics, the fusidanes. It is produced by fermentation under controlled conditions of the fungus Fusidium Coccineum.
-
The excellent distribution in various tissues, low degree of toxicity and allergic reactions and the absence cross-resistance with other clinically used antibiotics has made fusidic acid a highly valuable antibiotic,especially for skin and eye infections. The drug is used clinically both in its acid form, and as the sodium salt (Fusidin®), however Fusidin® is more favored one because of its better solubility in water, enabling a fast absorption from gastro-intestinal tract. As a result, it is more preferable to use sodium salt of fusidin in oral solid forms.
-
Fusidin® has the actions and uses of fusidic acid, and it has been shown that it ameliorates the course of several organ-specific immuno-inflammatory diseases such as chronic uveitis, Behcet’s disease, type I diabetes mellitus, Guillain-Barre syndrome, hepatitis, sepsis, pancreatitis, formalin-induced edema, multiple sclerosis, and scleroderma, whereby fucidin formulations have a great importance in pharmaceutical production.
-
Fusidin® can be presented in various formulations that differ significantly in their pharmacokinetic behaviors such as oral tablets, oral suspensions, intravenous formulations and topical preparation. Considering oral tablets, many of the early clinical studies were performed with capsule containing sodium fusidate. This was also the formulation marketed for many years in several countries. It is currently available as an oral tablet containing the sodium salt. Originally the sodium salt was available as an enteric-coated form but later it was reformulated as a film-coated tablet that appears to be better tolerated and gives higher blood levels.
-
Fusidic acid sodium salt was used in capsules as well as in tablets which were coated enterically. However by this enteric coating, the active fusidic acid sodium salt was not released before the tablets reached the part of the gastrointestinal tract in which the enteric coating would be dissolved. Depending on the time of passage through the stomach together with the food and the pH in the gastrointestinal tract, this led to unpredictable variations in the blood concentration of the patient undergoing treatment. Because of these adverse differences in blood concentration, the tablets without enteric coating were produced. Now, sodium fusidate is available in tablet, oral solution and injection form
-
PCT/WO9603128 A (LEO PHARMACEUTICALS PRODUCTS LTD. ET.AL.) describes the preparation of fusidic acid sodium salt tablets without an enteric coating by using dry granulation method in which a roller compactor was used. The compacted material so produced was size reduced to form a granulate having a bulk density in the range 0.45 to 0.9 g/m3 which was then formed into tablets.
FA was developed for clinical use in the 1960s and it is approved for human use outside of the United States, such as in the UK, Canada, Europe, Israel, Australia and New Zealand. It is typically prescribed at doses of 500 mg TID for treating skin and skin structure infections caused by Staphylococcus aureus (A. Bryskier,Fusidic Acid, Chapter 23, in Antimicrobial Agents: Antibacterials and Antifungals(Andre Bryskier, Ed., ASM Press, Washington, USA, 2005); Collignon et al., Int’l J. Antimicrobial Agents 12:S45-S58 (1999); D. Spelman, Int’l J. Antimicrobial Agents 12:S59-S66 (1999)), although some physicians have routinely prescribed the compound at 500 mg BID for treating skin and skin structure infections due to the long half-life of the compound (Fusidic Acid, in Principles and Practice of Infectious Diseases, 6th ed. (Mandell et al. eds., Elsevier, 2006)).
Treatment using FA has been well studied and it is generally regarded as safe when administered to humans, as evidenced by the fact that the drug has been in continuous use for more than 40 years. There are, however, several characteristics of FA that have prevented use of the drug against a wider spectrum of bacteria and in the treatment in additional types of infection. For example, approved dosing regimens have been shown to select for bacterial resistance, such as in S. aureus. Approved dosing regimens provide low multiples of the MIC and as a result, S. aureus resistant mutants can be selected after the first day of dosing. Once resistance has developed, FA is not effective against the resistant strains. Resistance is reported to occur if FA is used as a single drug as the resistance frequency at 4 and 8 times the MIC is in the range of 10−6 or 10−8 (Evans et al., J. Clin. Path. 19:555-560 (1966); Hansson et al., J. Mol. Biol.348:939-949 (2005), Jensen et al., Acta Pathol Microbiol Scand. 60:271-284 (1964); Besier et al., Antimicrob. Agents Chemo., 49(4):1426-1431 (2005); Gemmell et al., J. Antimicrobial Chemo. 57:589-608 (2006)).
The dosage of the drug cannot be simply increased as a means of avoiding development of resistance. It is difficult to achieve high concentrations of FA in the blood due to the substantial protein binding of the drug (approximately 95-97%) (K. Christiansen, International Journal of Antimicrobial Agents 12:S3-S9 (1999); Coutant et al., Diagn Microbiol Infect Dis 25:9-13 (1996); D. Reeves, J. Antimicrob. Chemo. 20:467-476 (1987); J. Turnidge, Int’l J. Antimicrobial Agents12:S23-S34 (1999); Rieutord et al., Int’l J. Pharmaceutics 119:57-64 (1995)). Moreover, high dosages of FA are not well-tolerated by patients receiving the drug. High doses of FA (e.g., 1 gram TID) are required if the drug is to be used in the treatment of bone and joint infections, less susceptible bacteria and other serious infections. However, treatment regimens using high doses of the drug induce nausea and vomiting and are rejected by patients (Fusidic Acid, inPrinciples and Practice of Infectious Diseases, 6th ed. (Mandell et al. eds., Elsevier, 2006); K. Christiansen, International Journal of Antimicrobial Agents 12:S3-S9 (1999); Nordin et al., Eur. J. Clin. Res. 5:97-106 (1994)).
In view of the tremendous costs associated with the de novo development of new anti-bacterials, expanding the indications for drugs that have already been demonstrated to be safe and effective is strongly needed. Overcoming the limitations on the uses of FA would broaden the population of bacterial infections against which it could be used and thus meet this need.
In a specific commercial pharmaceutical formulation, fusidic acid is presently marketed [see Monographs in the European Pharmacopeia 5.0] as a hemihydrate, which is the only hemihydrate form which has been described.
Patent GB 930,786 discloses salts of fusidic acid with organic and inorganic bases, solvates of fusidic acid, namely a benzene solvate and a methanol solvate. This patent further discloses an unspecified fusidic acid form with IR absorption bands (KBr) at 1265, 1385, 1695, 1730 and 3450 cm“1 and having a specific rotation [α]D 22 of minus 9 degrees (1% solution in CHCI3) obtainable by crystallisation of the methanol solvate of fusidic acid from ether. However, this form is distinct from the form of the present invention evident from the depicted IR spectrum in GB 930,786 which indicates that this form actually corresponds to the presently marketed hemihydrate form.
Solvates and salts of fusidic acid have also been disclosed in British patent GB 999,794. Patent ES 2208110 discloses two solvent free crystalline forms offusidic acid called Form I and Form II, and a crystalline hemihydrate called Form III which is identical to the presently marketed hemihydrate, respectively. The crystalline forms were identified and characterised by IR spectroscopy, differential scanning calorimetry, X-ray diffraction and melting points.
Patent WO 96/03128 discloses tablets containing a sodium salt form of fusidicacid and WO 86/03966 describes an ophthalmic gel composition comprising an undefined form of suspended fusidic acid.
Filed under: orphan drug status Tagged: anthony crasto, CEM-102, FUSIDIC ACID, GENERIC DRUG, medicinal chemistry, NEW DRUGS, Taksta, world drug tracker

































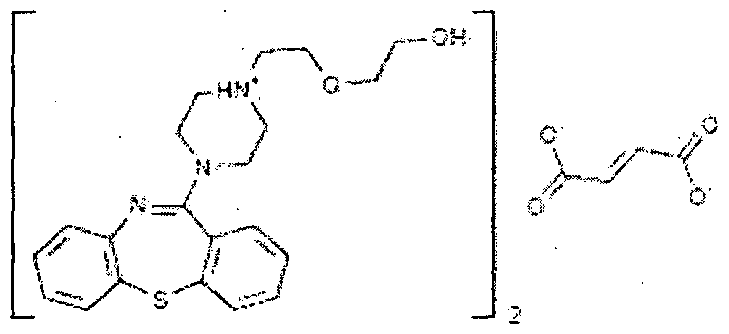
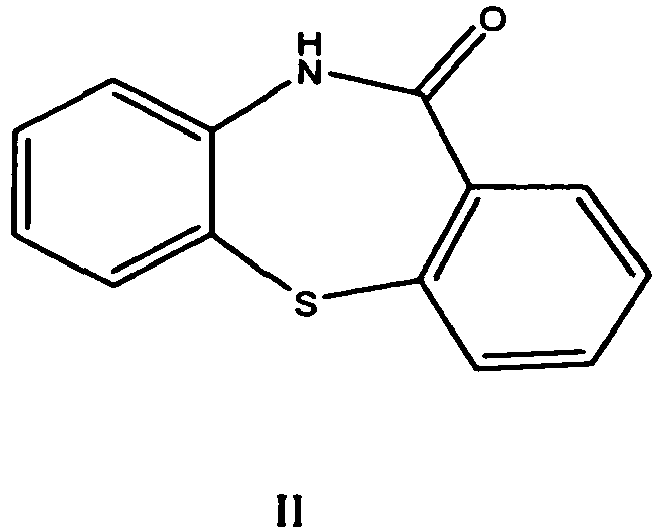


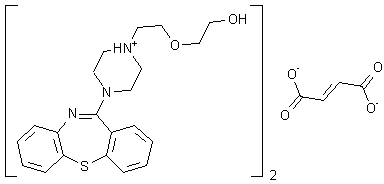











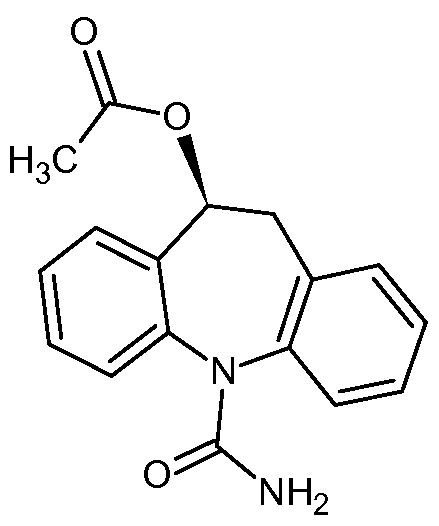
















































































.png)








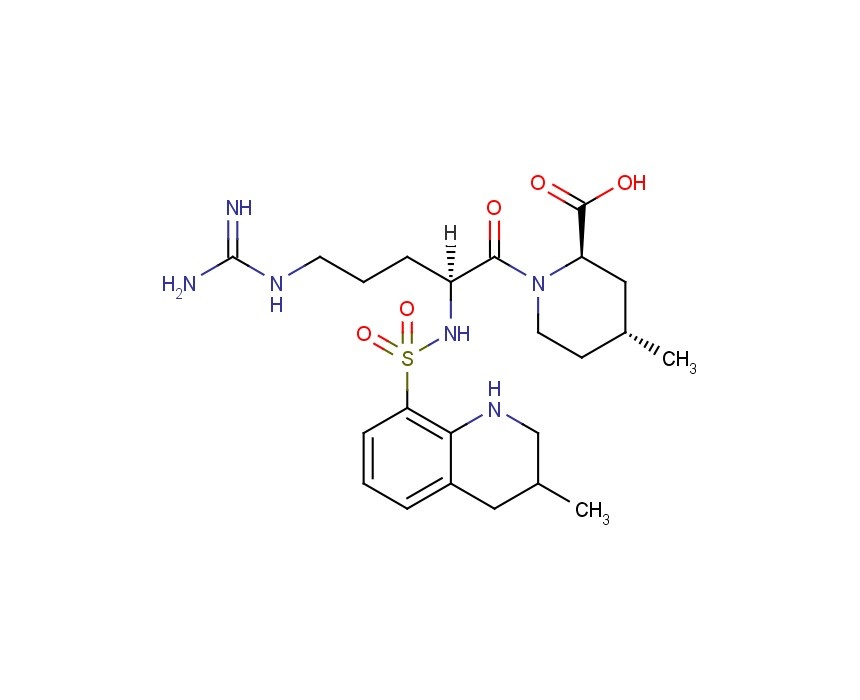













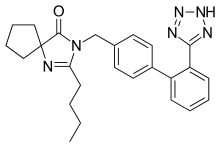













































 was prepared according to processes known in the art (e.g. U.S. Pat. No. 4,280,957) which comprise the basic hydrolysis of the corresponding ester.
was prepared according to processes known in the art (e.g. U.S. Pat. No. 4,280,957) which comprise the basic hydrolysis of the corresponding ester.




















































